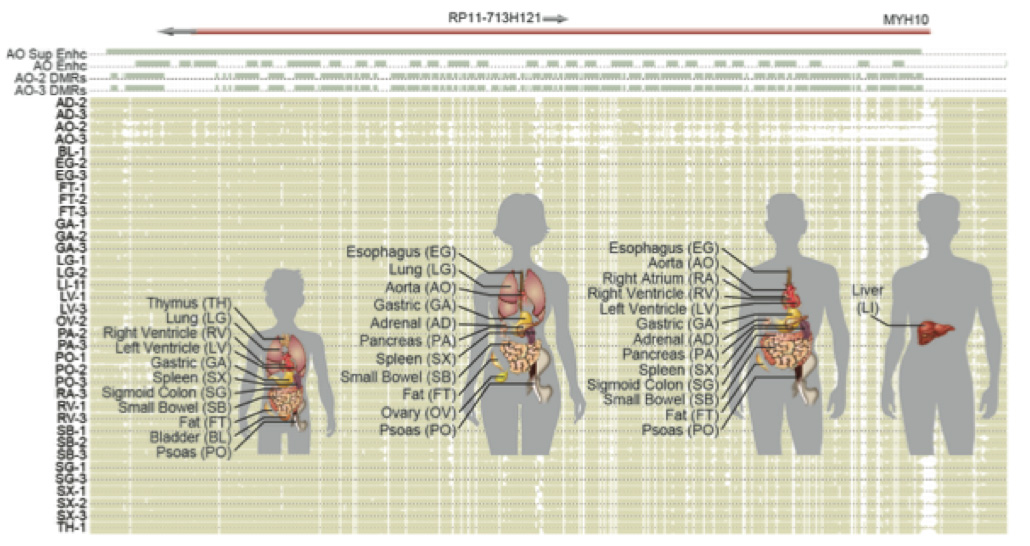
Understanding the diversity of human tissues is fundamental to disease and requires linking genetic information, which is identical in most of an individual’s cells, with epigenetic mechanisms that could play tissue-specific roles. Surveys of DNA methylation in human tissues have established a complex landscape including both tissue-specific and invariant methylation patterns. In this study, we report high coverage methylomes that catalogue cytosine methylation in all contexts for the major human organ systems, integrated with matched transcriptomes and genomic sequence. By combining these diverse data types with each individual’s phased genome, we identified widespread tissue-specific differential CG methylation (mCG), partially methylated domains, allele-specific methylation and transcription, and the unexpected presence of non-CG methylation (mCH) in almost all human tissues. mCH correlated with tissue-specific functions, and using this mark, we made novel predictions of genes that escape X-chromosome inactivation in specific tissues. Overall, DNA methylation in multiple genomic contexts varies substantially among human tissues.